
9th April 2023, Dr Chee L Khoo

One of the most important contributor to atherosclerosis is elevated cholesterol levels. Familial hypercholesterolaemia (FH) is a genetic disease that manifests as a disorder of cholesterol metabolism by mutations in hereditary genes usually in an autosomal dominant manner. Data suggest that 1 in 200 Caucasians are heterozygous for FH and that 1 in 160,000–300,000 are homozygous, which are much higher prevalence than those estimated a decade earlier (1). Yet in practice, we don’t seem to encounter too many of these patients. You can probably recall only a handful of patients with FH in your books. Is it that rare? The low numbers are a consequence of the gross underdiagnosis of the condition (2). Timely detection of FH is important because the presence of definite or probable FH increases 13 X risk of CAD compared to the general population [3]. The diagnosis should be made in primary care. And before the cardiovascular and cerebral events occur.
What is familial hypercholesterolaemia?
LDL-C binds onto its receptor on the surface of hepatocytes and is internalised and destroyed. The most common cause of FH, accounting for 80–85% of FH cases, is a mutation in the LDL-R gene leading to impaired LDL-R synthesis or impaired receptor binding (see Figure 1). LDL-C is not cleared from the circulation and blood LDL-C levels rises. Macrophages phagocytosed these lipoproteins and turn into foamy cells which are potent in causing atherosclerotic plaques. To date, >2000 mutant variants have been identified although only < 200 variants have been proven by complete in vitro functional studies to be causative of disease (4).
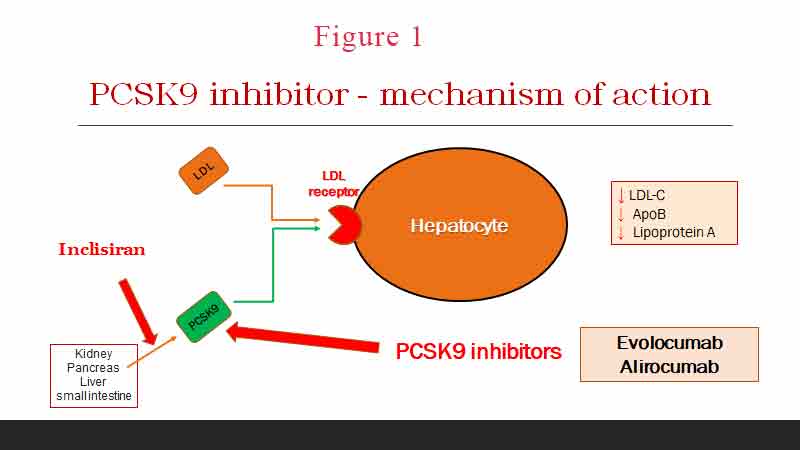
The second most common mutation is a mutation in APOB100, causing 5–10% of cases [24]. APOB is a ligand that encodes the one apolipoprotein included in LDL. Causative variants produce a protein that is unable to bind LDL-R. LDL-C is no longer cleared from the circulation.
Proprotein convertase subtilisin/kexin type 9 (PCSK9) is an enzyme encoded by the PCSK9 gene in humans on chromosome 1. It also binds to the LDL-C receptor on hepatocytes and reduces the clearance of LDL-C from circulation. PCSK9 variants can either be loss-of-function variants, generating less functional proteins, or gain-of-function variants, producing more active proteins. Both mechanisms lead to a reduced expression of LDL-R resulting in plasma LDL-C accumulation. Mutation in the PCSK9 gene is rare and accounts for about 2% of cases of FH.
How do we screen for FH?
A diagnosis of FH can be made based on clinical criteria or by DNA testing. Most patients with FH are asymptomatic. Physical examination may reveal cutaneous xanthomas in hands, toes, feet, elbows, knees and eye lids. Palpation of the heels may reveal thickening of the achilles tendons. The deposition of lipids in the cornea can cause single corneal circle, called arcus cornealis. Historical recall of joint pains may indicate past macrophage induced joint inflammation.
Calcified aortic valve is detected statistically more frequently in patients with FH. This pathology is observed in almost 100% of patients with homozygous FH. Patients with heterozygous FH mostly have subclinical form or aortic valve stenosis. Patients with FH predominantly does not have arterial hypertension, type 2 diabetes mellitus, metabolic syndrome, or hyperuricemia. The incidence of these diseases is statistically lower than in the population.
While genetic studies are available to confirm the presence of genetic mutation, access to these services are limited and the waiting lists for confirmation is typically long and primarily accessible in the major cities. The criteria of the Dutch Lipid Clinic (DLC) are the most widely used in clinical practice (8). Based on these criteria, FH is diagnosed referring to blood lipid levels, family history of IHD (early death from myocardial infarction or other early IHD), patient’s history of early coronary artery disease (CAD) and objective examination (tendon xanthoma, arcus cornealis, thickened achilles tendon). See Table 1.
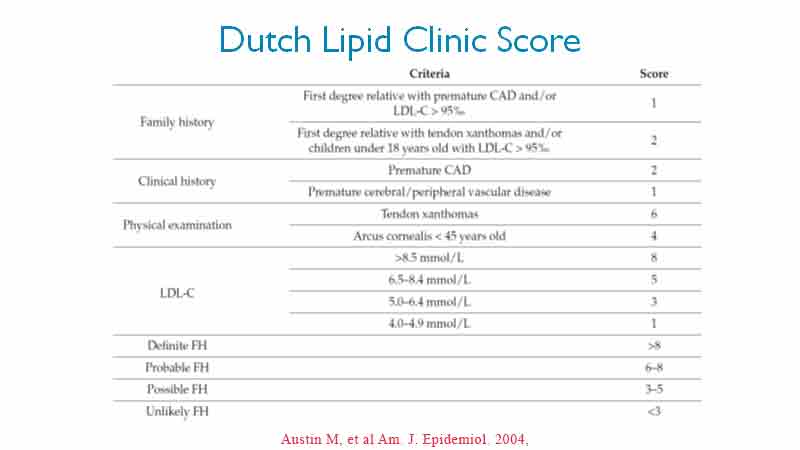
Remember, there are a lot more variants out there than we have genetic tests for. Some cases of FH are of polygenic origin or caused by mutations in genes that have not been identified yet. A probable diagnosis of FH according to the DLC Diagnostic Criteria is sufficient for a working diagnosis to begin treatment.
FH screening within primary care is considered cost-effective. However, which screening approach in primary care has not been established. Cascade screening, whereby family members are traced from an established FH index case, is more cost effective than any other screening strategy currently available and is recommended in NICE guidelines. Approximately half of the first degree relatives of an index case will be found to have the FH mutation.
The European Society of Cardiology (ESC) and European Atherosclerosis Society (EAS) recommends screening in adult patients with (5):
- TC >_8 mmol/L without treatment in an adult or adult family member
- Premature CHD (<55 y in men or <60y in women) in the patient or a family member;
- Tendon xanthomas in the patient or a family member; or
- Sudden premature cardiac death in a family member
Screening in children is recommended FH is diagnosed in children are based on similar criteria above. Testing during childhood is optimal to discriminate between FH and non-FH using LDL-C. In children with a family history of high cholesterol or premature CHD, an accepted cut-off is >4.0 mmol/L. If a parent has a known genetic defect, the diagnostic level for the child is >3.5 mmol/L. If possible, genetic testing of the child is suggested.
The more items from the DLC table that are present, the lower the LDL-C has to be before we suspect FH may be present. LDL-C cut off range from 4.0 mmol/L to 8.5 mmol/L.
It’s all about LDL-C levels, isn’t it?
It is insightful to look at the (UK) Simon Broome Register of Familial Hyperlipidaemia which began in 1980 (6). The cohort was followed up for 2234 person years during 1980-9. The excess mortality from this cause was highest at age 20-39 (~10X) and decreased significantly with age. There was no excess mortality by the time you get to 60-74 y group. The standardised mortality ratio for all causes was 1.83X and also was highest at age 20-39 (9X). There was no difference between men and women.
The Register presented its update in 2018 (7). They compared changes in CHD mortality in patients with heterozygous FH pre 1992, before lipid-lowering therapy with statins was used routinely, and in the periods 1992-2008 and 2008-2016. Overall, despite treatment with statins, there was a 2.4X excess mortality amongst patients with definite FH and 1.8X excess mortality amongst patients with probable FH. Even with patients with definite FH and established CAD, treatment reduced mortality from a 4.83X excess pre-1992 to 4.66X in 1992-2008 and 2.51X post-2008, while in women the corresponding values were 7.23X, 4.42X and 6.34X. This suggests that FH patients with pre-existing CHD and women with FH may not be treated adequately.
Remember, in patients with FH, the damage from elevated lipids begin from birth. Perhaps, much damage has already occurred before the first statin tablet was ever taken.
Guideline based treatment
It would seem that treatment of hypercholesterolaemia whether it is in patients with or without FH is pretty straight forward. However, like all lipid lowering therapy, the first question to ask is what the CV risk of the patient is. Remember, the burden of disease is high since these patients have hypercholesterolaemia since birth.
FH patients with no prior ASCVD or other major risk factors are automatically considered as high CV risk. FH patients with ASCVD or who have another major risk factor are treated as very-high-risk (5).
The next question is what the target LDL-C should be. In primary prevention, the ESC/EAS 2019 guidelines recommend a >50% reduction of LDL-C from baseline and a target LDL-C of < 1.8 mmol/L, though the lower the better (5). If this is not achievable by statin therapy alone, then the addition of ezetimibe is reasonable. If the goal LDL-C is still not achieved with maximally tolerated statin and ezetimibe, or the patient is statin intolerant, then PCSK9 inhibitor can be considered.
FH patients at very-high risk of ASCVD due to a prior history of ASCVD or another major risk factor, LDL-C goals are a >50% reduction of LDL-C from baseline and an LDL-C <1.4 mmol/L.
FH in pregnant women
Statins and other systemically absorbed cholesterol-lowering medications should be ceased three months before planned conception as well as during pregnancy and while breastfeeding (5). All women of childbearing age with FH should be offered pre-pregnancy counselling prior to starting treatment with statins.
FH in children
Treatment of children with FH includes a healthy lifestyle and statin treatment. A heart-healthy diet should be adopted early in life and statin treatment should be considered at 6-10 years of age. Statin treatment should be started with low doses and the dose should be increased to reach goals. The goal in children >10 years of age is an LDL-C <3.5 mmol/L and at younger ages a >50% reduction of LDL-C (5).
Other lipid lowering agents?
Other non-statin therapies such as niacin, bile acid sequestrants, fibrates, or apoB targeting therapies have very little evidence in FH. Emerging new LDL-C lowering therapies such as bempedoic acid (especially when given in combination with ezetimibe) and inclisiran appear promising.
In summary, FH is rare if we are not on a look out for it. The gold standard for diagnosis is genetic studies but not all variants have been fully identified and aetiology could be polygenic. The Dutch Lipid Score does well in identifying patients who have probable FH. Screening for FH can and should occur in primary care. Treatment needs to be initiated as soon as possible as the burden of disease commence from birth. Patients with FH have a 2.4- 10X excess mortality compared to the general population depending on how treatment was initiated early and aggressively or not.
References
- Singh, S.; Bittner, V. Familial Hypercholesterolemia—Epidemiology, Diagnosis, and Screening. Curr. Atheroscler. Rep. 2015, 17, 482. Available online: https://pubmed.ncbi.nlm.nih.gov/25612857/
- Nordestgaard BG,Chapman MJ, et al. European Atherosclerosis Society Consensus Panel. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European atherosclerosis society. Eur Heart J 2013, 34: 3478-3490a
- J. Slack, Risks of ischaemic heart-disease in familial hyperlipoproteinaemic states, Lancet 2 (7635) (1969) 1380e1382.
- Austin MA, Hutter CM, Zimmern RL, Humphries SE. Genetic causes of monogenic heterozygous familial hypercholesterolemia: a HuGE prevalence review. Am J Epidemiol 2004, 160(5): 407–420
- The Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and European Atherosclerosis Society (EAS). 2019 ESC/EAS Guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. European Heart Journal (2020) 41, 111[1]188. doi:10.1093/eurheartj/ehz455
- Risk of fatal coronary heart disease in familial hypercholesterolaemia. Scientific Steering Committee on behalf of the Simon Broome Register Group. BMJ. 1991 Oct 12;303(6807):893-6. doi: 10.1136/bmj.303.6807.893.
- Humphries SE, Cooper JA, Seed M, et al; Simon Broome Familial Hyperlipidaemia Register Group. Coronary heart disease mortality in treated familial hypercholesterolaemia: Update of the UK Simon Broome FH register. Atherosclerosis. 2018 Jul;274:41-46. doi: 10.1016/j.atherosclerosis.2018.04.040. Epub 2018 May 1.
- Austin, M.A.; Hutter, C.M.; Zimmern, R.L.; Humphries, S.E. Humphries, Familial Hypercholesterolemia and Coronary Heart Disease: A HuGE Association Review. Am. J. Epidemiol. 2004, 160, 421–429.