
11th July 2021, Dr Chee L Khoo

3 years ago we previewed the highly anticipated drug LY3298176, a novel dual GLP1 and GIP receptor agonist which activate both GLP1 and GIP receptors in islet cells when we look at twincretins. In healthy human subjects, LY3298176 caused weight loss and improved glucose tolerance. In subjects with T2D, LY3298176 reduced fasting glucose, glucose excursions and body weight with increasing doses. We examined the results of the 26-week randomised placebo and active comparator trial which compared LY3298176 with dulaglutide and placebo. LY3298176 resulted in remarkable jaw dropping reduction in HbA1c and body weight. 4 efficacy clinical trials have since been completed and its approval by the regulatory authorities will be imminent. Three more clinical trials are due to report soon.
Physiology of the incretins
GLP1
The two main incretin hormones are glucagon like peptide -1 (GLP-1) and gastric inhibitory peptides (GIP). GLP-1 is a 30 amino acid peptide secreted by L cells of the intestine. Once released into the circulation, GLP-1 binds to the GLP-1 receptor, which is expressed in the pancreas, gastrointestinal tract, kidney, heart and brain. Endogenous GLP-1 has a short half-life of 1–2 min and is metabolised by the enzyme DPP4. In additional to the well-known insulinotropic property in response to nutrients in the gut, GLP-1 also has a glucagonostatic effect (i.e. inhibit glucagon secretion) in the hyperglycaemic and normoglycaemic state but not during hypoglycaemia.
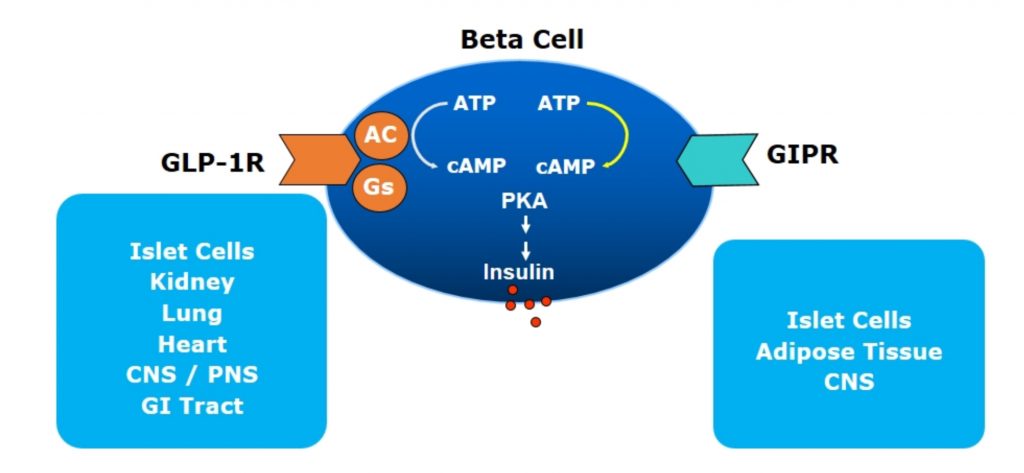
There are now 6 GLP1-RAs approved for use in Australia with a number of them on the PBS for the management of hyperglycaemia in type 2 diabetes (T2D). Both incretins bind to pancreatic β-cells (and other cells too – see below), both stimulate insulin release and both result in reducing plasma glucose. They work by binding to specific G protein-coupled receptor (GPCR) on the β-cell surface which activates cAMP leading to insulin release (see Figure 1). The GLP1 specific GPCR are present in islet cells (β, δ and possibly α-cells), CNS, GI Tract, kidneys, lungs and heart. The GIP specific GPCR, on the other hand, are found primarily in islet cells, CNS and adipose tissues.
GIP
What about the GIP? Why hasn’t it been used till now since it, too, binds onto β-cells and stimulate insulin secretion and thence, reduce glucose levels? GIP is a 4 amino acid peptide, produced by the K cells of the intestine, mainly located in the duodenum and proximal jejunum. GIP is released in response to oral nutrients, especially carbohydrates and lipids. GIP, like GLP-1, has a short half-life (4–7 min) and is inactivated by DPP4, shortly after release. GIP receptors are present in various tissues such as the pancreas, adipose tissue, gastric mucosa, heart, adrenal cortex, bone and brain. Unlike GLP-1, GIP has dual functions: a glucagonotropic property in the normoglycaemic and hypoglycaemic state, and glucagonostatic in the hyperglycaemic state. In other words, GIP causes glucagon release if glucose is low but backs off if glucose is high. Isn’t it convenient?
Incretins in T2D
The incretin effect is significantly reduced in patients with T2D. It is thought that the loss of incretin effect is from (1) a reduction in secretion of incretin hormone in response to nutrients (hyposecretion) and/or (2) a reduction in insulinotropic effect on pancreatic beta cells (insulin resistance). Early studies showed that there was hyposecretion of GLP-1 in people with T2DM, but emerging evidence does not support this observation. To counter the reduction in insulin sensitivity of the β-cells, supraphysiological doses of GLP1-RA (as in GLP1-RA therapy) partially restore the incretin effect.
With respect to GIP, there are consistent reports demonstrating no reduction in GIP secretion in people with T2D [1-3]. However, GIP resistance, a complete loss of insulinotropic effect of GIP at physiological concentrations as well as supraphysiological concentrations was observed in people with T2D. Very interesting though, some studies found that the glucagonotropic effect of GIP persists even in the hyperglycaemic state [4]. These observations in the past have made GIP unattractive as a therapeutic agent for T2D. However, emerging evidence suggests that GIP resistance can be modified and restored by improving glycaemic control. GIP receptors on pancreatic β-cells are downregulated owing to chronic hyperglycaemia, resulting in subsequent loss of insulinotropic effect. In other words, once we improve glycaemic levels, GIP doesn’t increase glucagon secretion.
Dual GIP and GLP1 agonist
Co-administration of GIP and a GLP1-RA in healthy individuals has an additive effect, generating a significantly increased insulin response compared with separate administration of each hormone [5,6]. Furthermore, the combined infusion produced a significant glucagonostatic effect while separate administration of GIP or GLP-1 did not suppress glucagon secretion more than glucose alone [6].
One of the limitations of the GLP1-RA therapy is the gastrointestinal (GI) side-effects (nausea, vomiting and diarrhoea) at high doses. Naturally, the higher the dose, the lower the glucose lowering is and the more body weight reduction is. The “average” weight loss for Saxenda 3mg daily is 10-11kg, Trulicity 1.5mg weekly is 4-5kg and Ozempic 1mg weekly is 6.5kg. We can push all the doses up higher but the dropout rate from GI side effects will increase. So, how do you create a dual agonist without the GI side effects?
First, you find a weak GLP1-RA. Then you add a GIP sequence to it. That’s exactly what they have done to create agent LY3298176 which is now called Tirzepatide. It was discovered by engineering GLP-1 activity into the GIP sequence. It is an imbalanced dual agonist in favour of GIP over GLP1-RA activity as the molecule shows equal affinity for the GIP receptors compared with native GIP but binds the GLP-1 receptors with 5-fold weaker affinity than native GLP-1. Weaker affinity for GLP1 receptors may mean less GI side effects. Tirzepatide also contains a C20 unsaturated di-acid acyl chain to affect albumin binding, and the overall properties of the molecule enable once-weekly dosing. Does it work in practice?
The Phase 3 clinical trials – SURPASS trials
- In SURPASS-1, all three tirzepatide doses (5,10,15mg) demonstrated statistically significant and clinically meaningful improvements in A1C and body weight reductions compared to placebo. Up to 92% of participants on tirzepatide achieved an A1C of less than 7%—the ADA’s recommended target for most people with diabetes. Up to 52% achieved an A1C of less than 5.7%—the level for people without diabetes.
- In SURPASS-2, all three tirzepatide doses delivered superior A1C and body weight reductions from baseline compared to semaglutide. Up to 92% of participants on tirzepatide achieved an A1C of less than 7% and up to 51% achieved an A1C less than 5.7%.
- In SURPASS-3, all three tirzepatide doses delivered superior A1C and body weight reductions from baseline compared to titrated insulin degludec. Up to 93% of participants on tirzepatide achieved an A1C of less than 7% and up to 48% achieved an A1C of less than 5.7%.
- In SURPASS-5, all three tirzepatide doses delivered superior A1C reductions and weight reductions compared to placebo both added to titrated insulin glargine. Up to 97% of participants on tirzepatide achieved an A1C of less than 7% and up to 62% of participants on tirzepatide achieved an A1C of less than 5.7%.
There are 3 further SURPASS trials yet to report. The SURPASS-CVOT trial will provide long-term data on the CV safety as well as efficacy of tirzepatide. SURMOUNT-1 is a phase 3, randomised, double-blind, placebo-controlled trial investigating the management of obesity in individuals without T2DM. SYNERGY-NASH is a randomized, double-blind, placebo-controlled phase 2 study comparing the efficacy and safety of tirzepatide in patients with non-alcoholic steatohepatitis (NASH).
Tirzepatide, a dual GIP/GLP-1 receptor agonist, is a new incretin-based therapy for type 2 diabetes. The degree of HbA1c reduction and weight reduction observed in Phase 3 clinical trials is unprecedented. Watch this space.
References:
- Vilsboll T, Krarup T, Sonne J, et al. Incretin secretion in relation to meal size and body weight in healthy subjects and people with type 1 and type 2 diabetes mellitus. J Clin Endocrinol Metab. 2003;88(6):2706–13.
- Calanna S, Christensen M, Holst JJ, et al. Secretion of glucose-dependent insulinotropic polypeptide in patients with type 2 diabetes: systematic review and meta-analysis of clinical studies. Diabetes Care. 2013;36(10):3346–52.
- Holst JJ, Windelov JA, Boer GA, et al. Searching for the physiological role of glucose-dependent insulinotropic polypeptide. J Diabetes Investig. 2016;7(Suppl 1):8–12.
- Chia CW, Carlson OD, Kim W, et al. Exogenous glucose-dependent insulinotropic polypeptide worsens post prandial hyperglycemia in type 2 diabetes. Diabetes. 2009;58(6):1342–9.
- Nauck MA, Bartels E, Orskov C, Ebert R, Creutzfeldt W. Additive insulinotropic effects of exogenous synthetic human gastric inhibitory polypeptide and glucagon-like peptide-1-(7–36) amide infused at near-physiological insulinotropic hormone and glucose concentrations. J Clin Endocrinol Metab. 1993;76(4):912–7.
- Elahi D, McAloon-Dyke M, Fukagawa NK, et al. The insulinotropic actions of glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (7–37) in normal and diabetic subjects. Regul Pept. 1994;51(1):63–74.
- Min T, Bain SC. The Role of Tirzepatide, Dual GIP and GLP-1 Receptor Agonist, in the Management of Type 2 Diabetes: The SURPASS Clinical Trials. Diabetes Ther. 2021 Jan;12(1):143-157. doi: 10.1007/s13300-020-00981-0. Epub 2020 Dec 15. PMID: 33325008; PMCID: PMC7843845.
- Willard FS, Douros JD, Gabe MB, Showalter AD, Wainscott DB, Suter TM, Capozzi ME, van der Velden WJ, Stutsman C, Cardona GR, Urva S, Emmerson PJ, Holst JJ, D’Alessio DA, Coghlan MP, Rosenkilde MM, Campbell JE, Sloop KW. Tirzepatide is an imbalanced and biased dual GIP and GLP-1 receptor agonist. JCI Insight. 2020 Sep 3;5(17):e140532. doi: 10.1172/jci.insight.140532. PMID: 32730231; PMCID: PMC7526454