
27th December 2020, Dr Chee L Khoo

Every time we come across some rare genetic disorder, we learn more about the intricacies of metabolism. Nothing is more true when it comes to lipid and lipoprotein metabolism. Familial combined hypolipidemia (FCHL) is a relatively recently recognised autosomal recessive disorder characterised by globally reduced levels of LDL and HDL cholesterol and triglyceride, with no apparent adverse effects. The gene for FCHL was found by screening families that appeared to have heterozygous familial hypobetalipoproteinemia (FHBL) but did not have the apoB mutation. The low LDL and HDL are caused by loss of function mutation affecting the gene encoding angiopoietin-like protein 3 (ANGPTL3). The discovery has opened up another target for treating hypercholesterolaemia despite maximal statins, ezetimibe and PCSK9 inhibitors.
Lipoprotein metabolism
Before we look at what ANGPTL3 is all about, we better revise our understanding of lipoproteins and their role in lipid metabolism. Lipoproteins consist of a cholesterol and triglyceride core surrounded by a phospholipid outer shell. The hydrophilic bits are oriented outwards towards the water-soluble plasma while the lipophilic bits are oriented inwards towards the lipid-soluble core. Embedded in the phospholipid shell are special proteins called apo-lipoproteins. Apolipoproteins give the outer shell some stability but more importantly identify the function of the lipoproteins. See Figure 1.
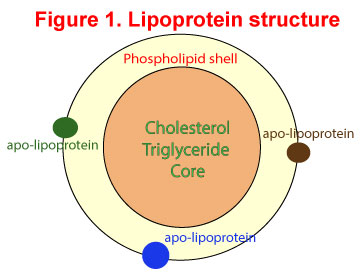
The primary function of lipoproteins is to transport molecules in plasma as fat cannot travel in water-soluble plasma. You will recall the common lipoproteins – chylomicrons, VLDL, LDL and HDL which cart cholesterol and triglycerides after digestion from the intestinal tract to the liver and from the liver to other organs. Apolipoprotein B (apoB) is the primary apolipoprotein in the chylomicrons, VLDL and LDL while apolipoprotein A (apoA) is the main apolipoprotein in HDL. The interaction of these apolipoproteins with enzymes in the blood, with each other, or with specific proteins on the surfaces of cells, determines whether triacylglycerols and cholesterol will be added to or removed from the lipoprotein transport particles. Used up lipoproteins then head back to the liver to be destroyed.
Abetalipoproteinemia (ABL) is caused by mutations in the gene encoding a subunit of the microsomal triglyceride transfer protein (MTTP). There are minimal or no apoprotein B‐containing lipoproteins circulating and patients are unable to absorb and transport fat and fat‐soluble vitamins. ABL is a rare, autosomal recessive disorder characterised by onset of malabsorption diarrhea soon after birth and slow development of a neurological syndrome thereafter. The neurological syndrome consists of ataxia, weakness of the limbs with loss of tendon reflexes, disturbed sensation, and retinal degeneration.
Familial hypobetalipoproteinemia (FHBL) is not as severe as ABL. There are some apoB lipoproteins but apoB and LDL-C levels are below the 5th percentile. Hypobetalipoproteinemia is not associated with malabsorption syndromes or neurological manifestations and leads to a reduced risk for cardiovascular disease. However, there are rare patients with homozygous hypobetalipoproteinemia who have mutations in both the apoB alleles and have plasma lipids similar to those in abetalipoproteinemia.
What is ANGPTL3?
It is a liver glycoprotein that reversibly inhibits lipoprotein lipase (LPL), which hydrolyse triglycerides within lipoproteins and clear chylomicrons and VLDL from circulation. It also inhibits endothelial lipase, which hydrolyses HDL. It has emerged as an important regulator of circulating cholesterol and triglycerides. In gain-of-function experiments in mice, increased ANGPTL3 activity led to increased total cholesterol and triglyceride levels. In the Dallas Heart study (1), loss-of-function mutations was reported in those in the lowest quartile of triglyceride levels. In other words, increased ANGPTL3 activity increase cholesterol and triglycerides and reduced ANGPTL3 activity reduce cholesterol and triglyceride levels.
Naturally, the discovery of the role of ANGPTL3 in lipid metabolism has open a therapeutic opportunity for treatment of refractory hyper-cholesterolaemia. Indeed, in a double-blind, placebo-controlled, phase 2 trial, patients with or without heterozygous familial hypercholesterolemia who had refractory hypercholesterolemia, with LDL cholesterol level ≥ 1.8 mmol/L with atherosclerosis or ≥ 2.5mmol/L without atherosclerosis were treated with evinacumab or placebo (2). Evinacumab is a fully human monoclonal antibody targeting ANGPTL3.
A total of 272 patients were randomised to received various doses of evinacumab (450mg weekly to 300mg subcutaneously fortnightly, 15mg/kg body weight to 5mg/kg body weight intravenously) or placebo. Nearly all the patients were receiving a PCSK9 inhibitor, 70% were receiving statin therapy (with 46% receiving a high-intensity statin), and 33% were receiving ezetimibe. Yet, the mean LDL cholesterol level at baseline was approximately 3.88 mmol/L, which suggests that many patients had untreated levels between 7.76 – 12.93 mmol/L. In the study, evinacumab significantly reduced the LDL cholesterol level, by more than 50% at the maximum dose. The incidence of serious adverse events during the treatment period ranged from 3 to 16% across trial groups.
Given that the action of evinacumab is independent of the LDL receptor and that the drug has effects on triglycerides and high-density lipoprotein cholesterol levels, in addition to the LDL cholesterol level, we await cardiovascular outcome trials with evinacumab.
Although approximately 30 million people have familial hyper-cholesterolemia worldwide, more than 90% of cases remain undiagnosed, and when the diagnosis is made, it is often made late and followed by suboptimal treatment (3-5). While our management options not so long ago were limited to statins, fish oils and ezetimibe, there is a promising pipeline of new therapeutic candidates in the horizon – therapies aimed at apoA and apoC3 and bempedoic acid (an agent that blocks cholesterol synthesis through inhibition of ATP citrate lyase).
Evinacumab is a new kid on the block in our battle against hyper-cholesterolaemia in patients on maximal therapy including PCSK9 inhibitors especially in patients with significant cardiovascular risks. No doubt, it will be a few years before it gets here and there will be significant restrictions on the PBS initially but these restrictions will loosen as outcome studies come online just as we see with the PCSK9 inhibitors (see last week’s article).
Did you know that you can refer patients to the Liverpool Genetic Clinic if you suspect they have familial hypercholesterolaemia? Here is the link.
References
1. Victor RG, Haley RW, Willett DL, Peshock RM, Vaeth PC, Leonard D, Basit M, Cooper RS, Iannacchione VG, Visscher WA, Staab JM, Hobbs HH; Dallas Heart Study Investigators. The Dallas Heart Study: a population-based probability sample for the multidisciplinary study of ethnic differences in cardiovascular health. Am J Cardiol. 2004 Jun 15;93(12):1473-80. doi: 10.1016/j.amjcard.2004.02.058. PMID: 15194016.
2. RS Rosenson, LJ Burgess, CF Ebenbichler, SJ Baum, ESG Stroes, S Ali, N Khilla, R Hamlin, R Pordy, Y Dong, V Son, D Gaudet. Evinacumab in Patients With Refractory Hypercholesterolemia. N. Engl. J. Med 2020 Dec 10;383(24)2307-2319
3. Sturm AC, Knowles JW, Gidding SS, et al. Clinical genetic testing for familial hypercholesterolemia: JACC Scientific Expert Panel. J Am Coll Cardiol 2018; 72: 662-80.
4. Singh A, Gupta A, Collins BL, et al. Familial hypercholesterolemia among young adults with myocardial infarction. J Am Coll Cardiol 2019; 73: 2439-50.
5. Duell PB, Gidding SS, Andersen RL, et al. Longitudinal low density lipoprotein cholesterol goal achievement and cardiovascular outcomes among adult patients with familial hypercholesterolemia: the CASCADE FH Registry. Atherosclerosis 2019; 289: 85-93.