
11th August 2024, A/Prof Chee L Khoo

Treating the CV risk factors like hypertension, diabetes mellitus, hypercholesterolemia and smoking have led to significant reductions in cardiovascular disease and mortality. However, up to 25% of first-time patients with myocardial infarction are SMuRF-less. We explored the issue last fortnight. Essentially, SMuRF-less patients are patients who do not have the usual standard modifiable risk factors (SMuRF). These are hypertension, diabetes, smoking and dyslipidaemia.
Hypertriglyceridaemia (HTG) is beginning to be looked at as one of those modifiable risk factors that may be responsible for the residual cardiovascular risks. Accumulation of triglyceride rich lipoproteins (TRLs) in plasma, impaired function of enzymes involved in TRLs lipolysis, and impaired hepatic clearance of cholesterol-rich TRLs remnants can lead to arterial deposition of TRLs and its remnants, promoting foam cell formation and arterial wall inflammation.
Epidemiological studies suggest that 10% – 25% of adults have elevated triglycerides (2,3). The prevalence varies by race, ethnicity, and ancestry (2,4). HTG is often underappreciated, underdiagnosed and undertreated.
Causes of hypertriglyceriaemia
HTG can either be primary or secondary. The primary causes are primarily from genetic mutations involving many genetic loci including LPL, apoC2, apoA5, LMF1, GPIHBP1, and GPD1. The genetic causes can be monogenic or polygenic. Familial chylomicronemia syndrome is a monogenic disorder caused by decreased clearance of chylomicrons often leading to triglycerides level > 20 mmol/L. Other monogenic mutations include transient infantile hypertriglyceridemia, congenital lipodystrophies, and familial dysbetalipoproteinaemia.
There are at least 300 loci influencing TG levels and these patients, expectedly, have high cardiovascular risks. Often, we can add secondary causes to these polygenic causes which further enhance the CV risks. Aboriginal and Torres Strait Islanders, people from Pacific Islands, Chinese, Southeast Asians and South Asians have additional secondary causes of high triglycerides as well higher propensity for central obesity.
The important implication of primary hypertriglyceridaemia is often, these patients have very high TG levels from a young age (if not from birth) and the burden of disease is high and these patients have a very high cardiovascular risk.
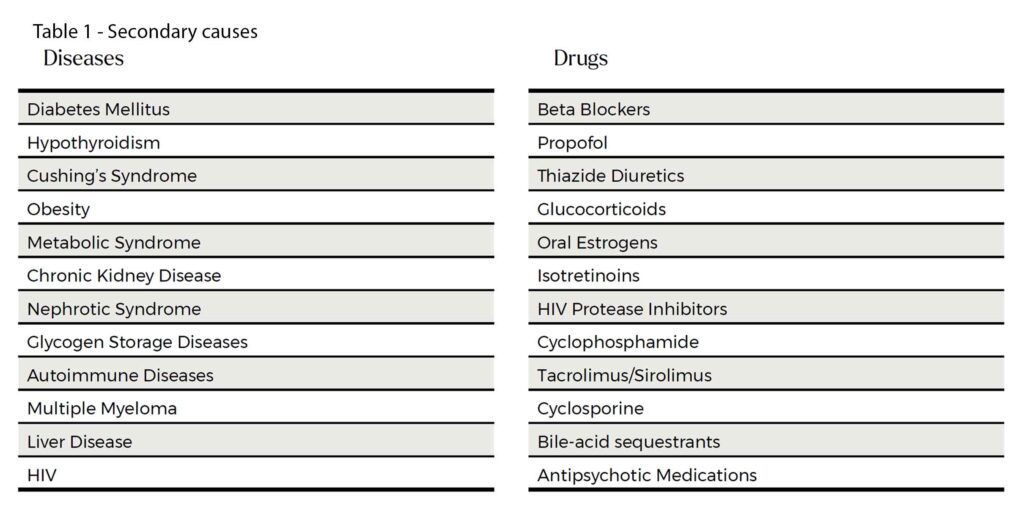
There are many secondary causes of hypertriglyceridaemia. See Table 1.
Degrees of hypertriglyceridaemia
Blood triglyceride concentrations can be measured in either fasting or non-fasting states, as both fasting and non-fasting samples correlate with cardiovascular risk but if the TG level is >4.5 mmol/L, a repeat fasting level should be organised. Unfortunately, the reference range from your friendly pathology providers can be confusing. The 2021 American Cardiology College (ACC) expert consensus statement categorise the degrees of HTG (5):
- Mild to moderate 1.7 – 5.6 mmol/L
- Severe >5.6 mmol/L
It is noted that cardiovascular events can occur in patients with triglycerides in the “normal range”. Both the ACC and the ESC do reference the optimal fasting triglyceride level as < 1.2 mmol/L (6).
Triglyceride Metabolism
Triglycerides comes from:
1) Exogenous TG synthesised by intestinal epithelial cells from the absorption of digested dietary fat. Exogenous TG is assembled with other lipids in intestinal cells and secreted into chylomicrons (CM) for transporting to the peripheral tissues.
2) Endogenous TG synthesised by hepatic uptake and utilization of circulating free fatty acids (FFA). These FFA are sourced from through various pathways, including the uptake of chylomicron residues, de novo lipogenesis (DNL) where glucose and other non-lipid precursors are converted to fatty acids, uptake of non-esterified fatty acids in plasma. The liver transports the TG to the peripheral tissues via very low density lipoproteins (VLDL).
The TRLs are thus, composed of CM and VLDL. TRLs travel to the peripheral tissues where their triglycerides undergo hydrolysis by lipoprotein lipase (LPL). LPL exist in a complex with glycosylphosphatidylinositol high density lipoprotein-binding protein-1 (GPIHBP1) on the surface of endothelial cells. The hydrolysis releases free fatty acids for use by the peripheral tissues of the body.
After triglycerides are stripped from chylomicrons and VLDLs, what is left behind are chylomicron and VLDL remnants. CM remnants are cleared by the liver while the VLDL remnants are further converted into LDL and HDL in the liver. Not all the CM remnants are cleared by the liver and similarly, not all the VLDL is converted by the liver. The remaining remnants remain in plasma and contribute to the atherosclerosis process.
In the liver, CM remnants exchange TG for cholesterol esters with HDL-C. This process involves apoCIII which is also involve in the liver uptake of VLDL. HTG disrupts the action of apoCIII which inhibits the production of HDL and uptake of VLDL.
Several modulating proteins have been identified which either inhibit (apolipoprotein CIII, angiopoietin-like protein 3 (ANGPTL3), angiopoietin-like protein 4 (ANGPTL4)) or augment the action of LPL (apolipoprotein AV). Inhibiting LPL raises TRL concentrations by limiting triglyceride-rich lipoprotein catabolism. Novel therapeutic agents which target the modulating proteins are currently in development for patients with elevated triglycerides. Many also improve other lipid related biomarkers such as LDL-C or apoB.
TG and CVD – what’s new
In recent years, a series of genetic, epidemiological, molecular biological and clinical investigations have unveiled a robust association between serum TG and TRLs levels and the residual risk of ASCVD. TRLs remnants may contain 5-20 times more cholesterol than LDL-C and therefore, more atherogenic than LDL-C. Not surprisingly, these remnant particles, are independently predictive of increased CV risk. TRLs can quickly penetrate the endothelial layer and they are prone to subendothelial accumulation. The more efficient the lipolysis of the TRL is, the more rapid removal of these atherogenic lipoprotein is.
Lipoproteins containing ApoB with a diameter less than 70 nm, which include CM remnants, VLDL, VLDL remnants, IDL, and LDL, can efficiently traverse the intact endothelium, allowing products from TRLs metabolism to accumulate beneath the vascular endothelium (7). These products sequestered within the arterial wall are pro-inflammatory attracting monocyte migration from the endothelial space to the subendothelial space where they differentiate into macrophages. Of course, when the macrophages ingest the lipoproteins, they become foam cells. These cholesterol-rich foam cells promote plaque formation. Macrophages and foam cells further secrete chemicals which promote TRL and TRL remnant retention. See Figure 1.
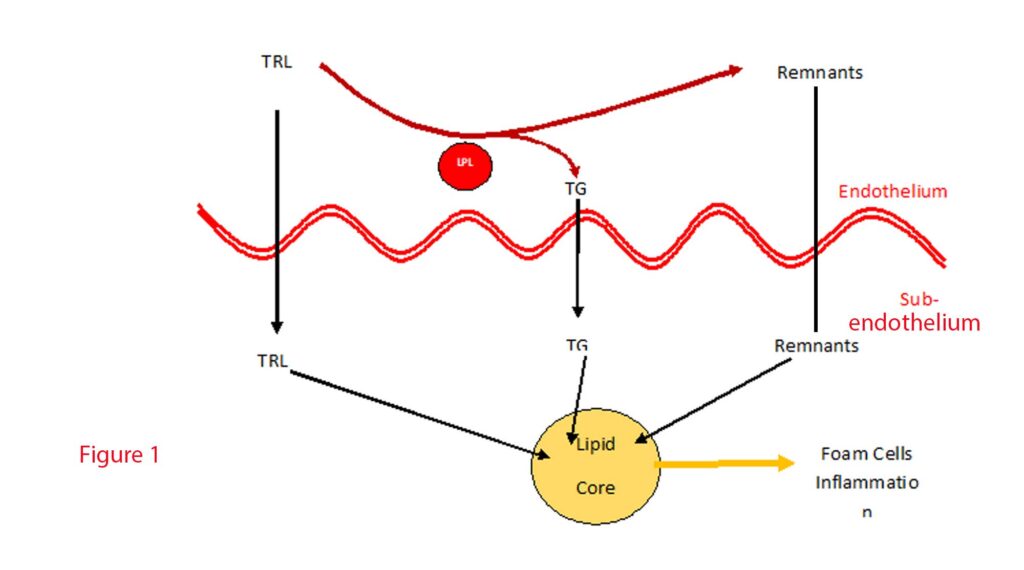
Over-accumulation of lipoproteins induces apoptosis of the macrophages which releases remnants and debris leading to necrotic cores within plaques termed “fragile plaques,” susceptible to rupture and thrombus formation, precipitating acute cardiovascular events (8).
TRLs accumulation in the arterial wall stimulates smooth muscle cells (SMCs) proliferation and inflammation by activating genes involved in SMC proliferation and inflammation regulation (9).
In summary, hypertriglyceridaemia is not a minor player in the pathogenesis of atherosclerotic cardiovascular disease. It may be one of the missing links in residual cardiovascular risks after the standard modifiable risk factors have been controlled. HTG not just augments existing risk factors especially elevated LDL-C but also directly contributes to the subendothelial lipid core, the subendothelial pro-inflammatory milieu, the vascular smooth muscle remodelling and increase the risk of acute plaque rupture.
In the next issue of GPVoice, we will explore what agents are available to manage HTG and the evidence behind their efficacy. More importantly, we will explore when to use them in our patients with HTG when we examine the newly released “A Consensus Statement of the International Atherosclerosis Society”.
References:
- https://athero.org/resources/triglycerides-revisited/. Accessed 11/8/2024
- Aggarwal R, Bhatt DL, Rodriguez F, Yeh RW, Wadhera RK. Trends in Lipid Concentrations and Lipid Control Among US Adults, 2007-2018. JAMA. 2022;328:737–745.
- Fan W, Philip S, Granowitz C, Toth PP, Wong ND. Hypertriglyceridemia in statin-treated US adults: the National Health and Nutrition Examination Survey. J Clin Lipidol. 2019;13:100–108.
- Gill PK, Dron JS, Dilliott AA, McIntyre AD, Cao H, Wang J, Movsesyan IG, Malloy MJ, Pullinger CR, Kane JP, Hegele RA. Ancestry-specific profiles of genetic determinants of severe hypertriglyceridemia. J Clin Lipidol. 2021;15:88–96.
- Virani SS, Morris PB, Agarwala A, Ballantyne CM, Birtcher KK, Kris-Etherton PM, Ladden-Stirling AB, Miller M, Orringer CE, Stone NJ. 2021 ACC Expert Consensus Decision Pathway on the Management of ASCVD Risk Reduction in Patients With Persistent Hypertriglyceridemia: A Report of the American College of Cardiology Solution Set Oversight Committee. J Am Coll Cardiol. 2021;78:960–993.
- Visseren FLJ, Mach F, Smulders YM, et al. ESC Scientific Document Group. 2021 ESC Guidelines on cardiovascular disease prevention in clinical practice: Developed by the Task Force for cardiovascular disease prevention in clinical practice with representatives of the European Society of Cardiology and 12 medical societies With the special contribution of the European Association of Preventive Cardiology (EAPC). Eur Heart J. 2021;42:3227–3337.
- Borén J, Chapman MJ, Krauss RM, Packard CJ, Bentzon JF, Binder CJ, et al. Low density lipoproteins cause atherosclerotic cardiovascular disease: pathophysiological, genetic, and therapeutic insights: a consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J. (2020) 41:2313–30. doi: 10.1093/eurheartj/ehz962
- Virmani R, Burke AP, Kolodgie FD, Farb A. Vulnerable plaque: the pathology of unstable coronary lesions. J Interv Cardiol. (2002) 15:439–46. doi: 10.1111/j.1540-8183.2002.tb01087.x
- Bermúdez B, López S, Pacheco YM, Villar J, Muriana FJ, Hoheisel JD, et al. Influence of postprandial triglyceride-rich lipoproteins on lipid-mediated gene expression in smooth muscle cells of the human coronary artery. Cardiovasc Res. (2008) 79:294–303. doi: 10.1093/cvr/cvn082