
1st August, 2018, Dr Chee L Khoo
When a diagnosis is idiopathic, it often means we don’t know its pathogenesis well. Often, a mixed bag of conditions is thrown in there because we are rather imprecise in defining them. Fibrosis is the final common pathway of lung injury and idiopathic pulmonary fibrosis used to have a mixture of fibrotic conditions lumped together under the umbrella diagnosis. As we know more about the pathogenesis of the different fibrotic conditions, we can now somewhat divide up the various interstitial pneumonias.
The American Thoracic Society classify idiopathic interstitial pneumonias (IIP) into 4 categories – chronic fibrosing IIPs, acute/subacute IIPs, smoking related IIPs and rare IIPs [1]. Of the fibrosing IIPs, IPF and fibrotic non specific interstitial pneumonitis (NSIP) are the most common.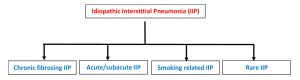
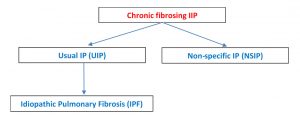
The radiological picture is interstitial pneumonia (IP) and after excluding other mimickers (see below), it becomes idiopathic pulmonary fibrosis (IPF). So, you will often term as UIP/IPF. Differentiating between the UIP and NSIP can be difficult because of overlapping clinical, pathologic and radiological presentation.
To complicate matters further, chronic hypersensitive pneumonitis (CHP) and stage 4 sarcoidosis (S4) are not part of the IIP but are differential diagnoses because they are common and share many features with the IIPs. Their prognoses are different and their treatment are very different.
Diagnosis is primarily clinical and radiological. Only very occasionally is tissue biopsy required.
IPF
It frequently occurs in the elderly male population (median age, 66 years). Risk factors for disease include cigarette smoking and gastroesophageal disease. Most patients with IPF demonstrate a gradual worsening of lung function over years, whereas some patients experience episodes of acute respiratory worsening despite previous stability (ie, acute exacerbation). Clinically, we find posterior lung basal crepitations on examination.
High resolution CT (HRCT) of the lungs reveals a variegated pattern of alternating areas of normal or near-normal lung with temporal heterogeneity of fibrosis consisting of scattered fibroblastic foci in the background of dense acellular collagen and honeycombing. Upper lobe–predominant fibrosis is not typical of UIP and is more common with hypersensitivity pneumonitis and sarcoidosis.
Pulmonary function tests demonstrate restrictive physiology with diminished Diffusion Capacity of Lungs for Carbon Monoxide (DLCO) and Forced Vital Capacity (FVC).
It used to be believed that UIP/IPF was driven by uncontrolled inflammation. Anti-inflammatory medications (prednisone, azathioprine, N-acetylcysteine, interferon-γ, etc.) were previously administered or tested in patients with UIP/IPF. They are no longer recommended as their efficacies are not proven. In 2014, 2 antifibrotic medications, nintedanib and pirfenidone, were approved in the United States for the treatment of IPF. Pirfenidone is an antifibrotic drug that reduces lung fibrosis through down-regulation of the production of growth factors and procollagens I and II. It was shown to reduce the rate of FVC decline as well as a statistically significant improvement in progression-free survival in the Assessment of Pirfenidone to Confirm Efficacy and Safety in Idiopathic Pulmonary Fibrosis (ASCEND) trial,[2,3].
Nintedanib is a tyrosine kinase inhibitor, originally developed as an anti-vascular agent for oncology indications, was tested for efficacy against IPF in several trials and these showed a significant impact in rate of FVC decline [4,5].
UIP/IPF has a poor prognosis with a mean life expectancy of 3.8 years. The American Thoracic Society (ATS) has provided guidelines for the accurate diagnosis of IPF. This is a diagnosis of exclusion.
NSIP
NSIP is most common among women in their 40s to 50s and nonsmokers.
NSIP is often a finding in patients with other connective tissue disease. Obtaining a comprehensive panel of serum autoantibodies and inflammatory markers, including but not limited to antinuclear antibody, rheumatoid factor, anti-Scl-70, antisynthetase antibodies (myositis panel), anti-Ro (SS-A), anti-La (SS-B), antiribonucleoprotein, aldolase, creatine kinase, erythrocyte sedimentation rate, anticyclic citrullinated peptide, and C-reactive protein, is crucial.
NSIP and UIP are both lower lobe–predominant diseases; however, NSIP is distinctively different radiographically from UIP because of its homogeneity and its subpleural sparing.
If progression of disease is seen, treatment with immunosuppressants is thought to be beneficial
IPF also needs to be differentiated from chronic hypersensitive pneumonitis (CHP) and stage 4 sarcoidosis (S4).
CHP
CHP is an interstitial lung disease in genetically predisposed individuals caused by an exaggerated immune response to chronic inhalation of a variety of antigens in the environment (fungal, bacterial, protozoal, and animal proteins, or low-molecular-weight chemical compounds, etc.). With long-term inflammation, CHP with progressive fibrosis and bronchiolitis obliterans may develop.
A careful history regarding the occupational/domestic environment and hobbies is crucial (ie, bird keeping, hay feeding, feather duvet and pillows at home, air conditioning, contaminated ventilators in the buildings, and formation of mold on room walls or in brake fluid oils, or within wind instruments).
Radiologically, CHP is different from UIP radiographically mainly because instead of being peripheral, it is an airway-centered disease, which makes sense given the way in which it is acquired. Second, CHP is upper-lobe predominant and unlike UIP, which is lower-lobe predominant
Stage 4 Sarcoidosis
Sarcoidosis is a multisystemic inflammatory disease of unknown aetiology, characterized by the presence of noncaseating granulomas, and predominantly affecting lung. Approximately 20% of patients develop pulmonary fibrosis (ie, radiographic stage 4 sarcoidosis) with substantially increased mortality, therefore it can be another mimicker of UIP/IPF. Sarcoidosis has more in common radiographically with NSIP and CHP than with UIP because the fibrosis is not peripheral but instead follows the broncho-vascular bundles. Its upper-lobe predominance helps to differentiate S4 from NSIP. A posterior predominance and absence of air trapping helps to differentiate S4 from CHP, which is upper lobe but more frequently anterior.
- Reference
Travis WD, Costabel U, Hansell DM, et al. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med 2013;188:733–48 - Raghu G, Anstrom KJ, King TE Jr, Lasky JA, Martinez FJ. Prednisone, azathioprine, and N-acetylcys-teine for pulmonary fibrosis. N Engl J Med 2012; 366:1968–77.
- Noble PW, Albera C, Bradford WZ, et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): Two randomised trials. Lancet 2011; 377:1760–9.
- Richeldi L, Costabel U, Selman M, et al. Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N Engl J Med 2011;365:1079–87.
- Richeldi L, du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014;370:2071–82.
- Pollack A. F.D.A. approves first 2 drugs for treatment of a fatal lung disease. NY Times. October 14, 2014.