
11th February 2024, A/Prof Chee L Khoo

Hypertriglyceridaemia (HTG) often accompanies hypercholesterolaemia but that is often ignored as it is considered to be a minor villain. Older clinical trials on lipid lowering agents excluded subjects with HTG and that is partly why the significance of HTG is often ignored. The interplay between HTG and atherosclerotic cardiovascular disease (ASCVD) is real but extremely complicated. The management of HTG is even more complicated. Epidemiological studies in the general population consistently demonstrate a strong, positive association of plasma triglyceride levels with risk of ASCVD but it is not easy to remove the confounding effects of changes to lipoproteins and the reduced HDL-C when plasma triglycerides are high. It might be insightful to explore the metabolism, causes and consequences of HTG in our patients.
Triglyceride (TG) itself is not found in the atherosclerotic plaque. Thus, the influence of HTG on ASCVD is likely to be indirectly through the impact of HTG on the metabolism of cholesterol-carrying lipoproteins and possibly, by activation of inflammatory pathways. So, let’s have a look at those cholesterol-carrying lipoproteins and their relationship with HTG.
Triglycerides and Lipoproteins
Apolipoprotein B (apoB) is the primary apolipoprotein of chylomicrons, VLDL, IDL, LDL and lipoprotein a particles. ApoB on the LDL particle acts as a ligand for LDL receptors. There are 2 main isoforms, apoB48 and apoB100. Both share a common N-terminal sequence, but apoB48 lacks apoB100’s C-terminal LDL receptor binding region. Actually, apoB48 is so-called because it constitutes 48% of the sequence for ApoB100.
From systematic meta-analyses of LDL cholesterol (LDL-C) lowering studies, for each 1.0 mmol/l drop in LDL-C decrease, there is a 22% relative reduction in ASCVD risk (1). Currently, our measurement of LDL-C is an estimate or calculation from a formula based on total cholesterol, TG and HDL-C which really doesn’t measure the real LDL-C concentration as it is affected by TG and HDL levels. Now, because there is one apoB100 molecule per LDL-C, we can accurately quantify the LDL-C concentration by measuring apoB100 concentration. In fact, we can now do that using nuclear magnetic resonance spectroscopy. Not surprisingly, it is well established that apoB100 levels are associated with coronary heart disease. They are a far better predictor of ASCVD than LDL-C concentrations (2,3).
The optimal plasma triglyceride concentration appears to be in the range below 1.2 mmol/L (4,5). As plasma TG rises above this level, abnormalities in VLDL and LDL begin to occur. VLDL is the vehicle to deliver TG from the liver to other organs. VLDL rises when plasma TG rises.
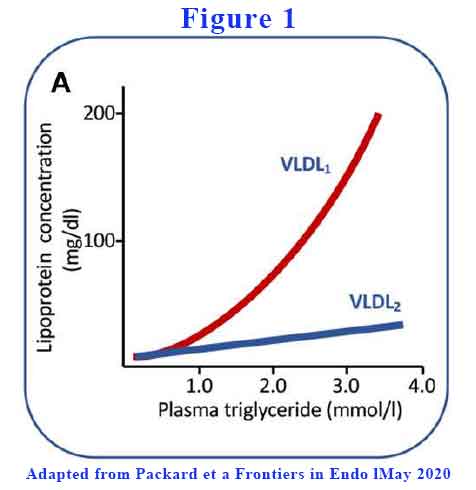
The liver can vary the amount of lipid it loads into the VLDL. Large TG-rich VLDL (VLDL1) have a diameter range of 50–80 nm and consist of about 70% triglyceride by mass. Smaller and less TG-rich VLDL (VLDL2) are 30–50 nm in diameter and consist of about 30% triglyceride. As plasma TG rises above 1.2 mmol/L, total VLDL rises with VLDL1 a lot more than VLDL2 (see Figure 1).
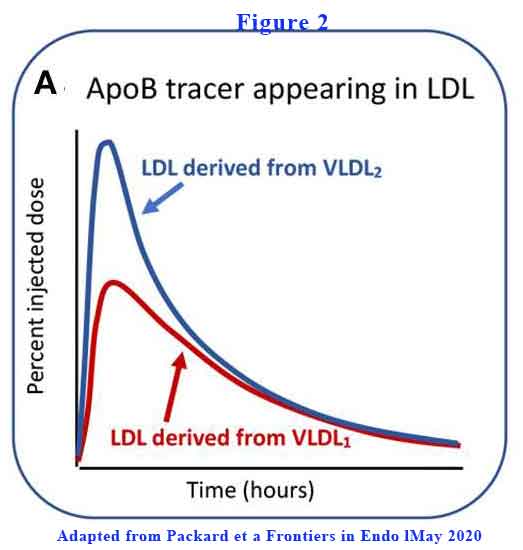
VLDL2 is more rapidly converted to LDL-C than VLDL1 (see Figure 2).
Further, VLDL1has a prolonged residence time in circulation. VLDL1 once secreted from the liver enters a delipidation cascade leading to the formation of smaller VLDL2, IDL, and LDL (see Figure 3).
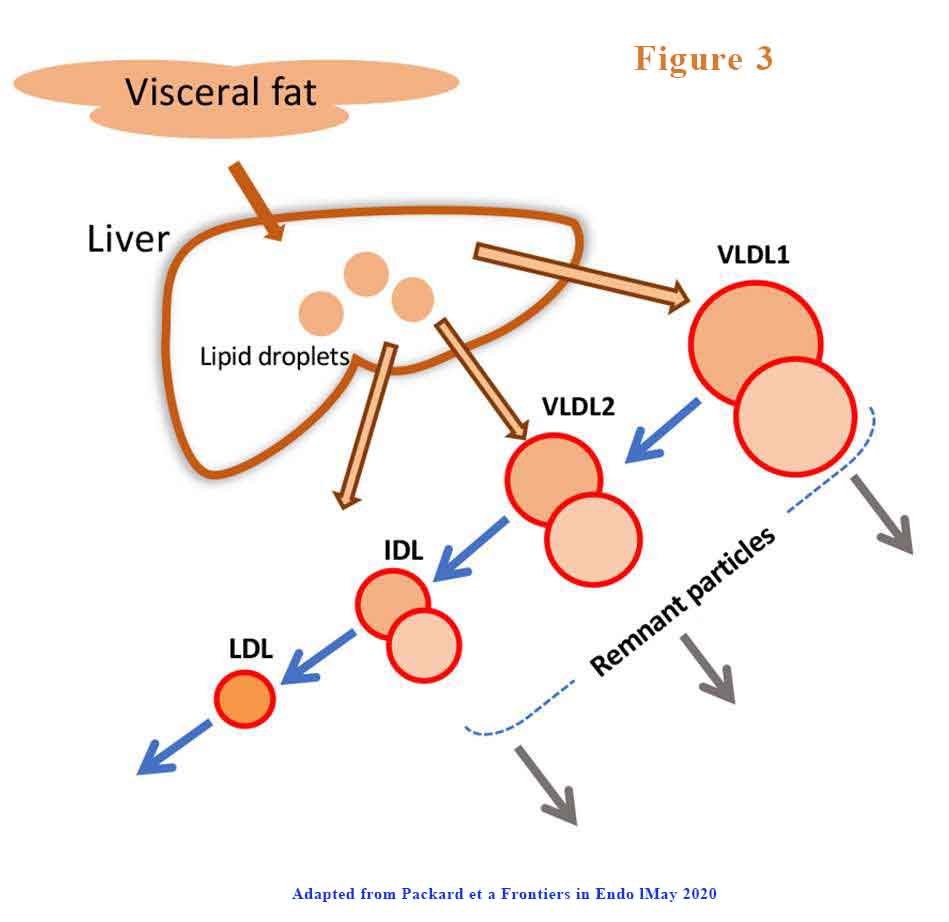
The proportion of VLDL1 and VLDL2 that is produced or degraded changes. The products of inefficient and incomplete VLDL delipidation are considered “metabolic remnant particles”. They accumulate in proportion to the increase in plasma triglyceride and are believed to be able to contribute to the deposition of cholesterol in atherosclerotic lesions as well as promote inflammatory processes.
The factors that drive VLDL1 and VLDL2 synthesis are different. VLDL1 synthesis is driven by the supply of triglyceride from intracellular stores, de novo lipogenesis, fatty acid uptake, and hepatic chylomicron remnant removal (6,7). Overproduction of smaller VLDL2, on the other hand, is linked to raised cholesterol levels (8) and is a feature of familial hypercholesterolemia (9,10).
Causes of HTG
There is a general increase in plasma triglyceride (TG) as we get older. This rise is driven, at least in part, by the age-related increase in body weight and adiposity since there is a strong link between being overweight or obese and higher rates of hepatic triglyceride synthesis and VLDL secretion. We know that weight reduction leads to TG lowering and a decrease in the production of VLDL. TG also rise in pregnancy likely as a result of an action of oestrogen on VLDL assembly and secretion in order to deliver triglyceride to the placenta.
The liver uses various lipoproteins to deliver TG and cholesterol to peripheral tissues. It is also the means to regulate the amount of lipids stored in the liver. Thus, when intrahepatic fat increases (as in non-alcoholic fatty liver disease (MASLD)), there is increased VLDL, apoB and TG synthesis.
Not surprising, hormones related to glucose and lipid homeostasis will impact on TG production and clearance. Insulin resistance is positively associated with VLDL1 synthesis rates in both normoglycemic and type 2 diabetic subjects while glucagon acts to suppress VLDL triglyceride release . In patients with type 2 diabetes, VLDL- triglyceride and-apoB production rates are positively related to the extent of hyperglycaemia (even in well-controlled patients).
Naturally, excessive calorie intake leads to higher TG storage in adipose and other tissues. Of the major dietary constituents, carbohydrate intake seems to have the greatest impact on plasma triglyceride levels. Ingestion of a high carbohydrate diet leads to the development of raised VLDL due to an increase in VLDL-triglyceride and -apoB production rates and a decrease in clearance. A carbohydrate-restricted diet leads to rapid, beneficial changes in hepatic lipid metabolism with decreased lipogenesis, increased fatty acid oxidation, and substantial reductions in VLDL triglyceride and plasma apoCIII levels.
Consequences of HTG
It is known that chylomicrons and VLDL compete for the same lipolytic mechanism with the former being preferred. When chylomicrons appear in circulation, apoB100 levels in VLDL rises sharply . Clearance rates of both apoB48 and apoB100 particles in VLDL1 and VLDL2 were retarded markedly in hypertriglyceridaemic subjects with residence times rising from 1 to 4 h in individuals with low triglyceride (<1.2 mmol/l) to 4–13 h in those with triglyceride >2 mmol/l (11). It follows, therefore, that both liver and intestine are responsible for the generation of long-lived remnant particles that could promote atherogenesis in hypertriglyceridaemic subjects.
The presence of insulin resistance is positively associated with VLDL1 synthesis rates in both normoglycemic and type 2 diabetic subjects while glucagon acts to suppress VLDL triglyceride release (12, 13). In patients with type 2 diabetes, it can be seen that VLDL- triglyceride and-apoB production rates were positively related to the extent of hyperglycaemia (even in well-controlled patients). Insulin administered acutely to normal and diabetic subjects; the former showed suppression of VLDL1 (but not VLDL2) production while the latter showed no such effect (13).
Thus, we can see that TG metabolism is closely linked to lipoprotein metabolism. It is not a minor villain. HTG has a major impact not just on the atherogenic VLDL and LDL concentration but also their direct effects on the atherosclerotic plaque. While diabetes and insulin resistance often lead to HTG, the effects cuts both ways. In a large-scale prospective cohort study of young adults, cumulative exposure to hypertriglyceridemia was significantly associated with an increased risk of type 2 diabetes, independent of lifestyle-related factors. We need to target both hypercholesterolaemia as well as HTG in patients with or without diabetes.
References:
1. Baigent C, Blackwell L, Emberson J, Holland LE, Reith C, Bhala N, et al. Efficacy and safety of more intensive lowering of LDL cholesterol: a metaanalysis of data from 170,000 participants in 26 randomised trials. Lancet (2010) 376:1670–81. doi: 10.1016/S0140-6736(10)61350-5
2. Cromwell WC, Otvos JD, Keyes MJ, Pencina MJ, Sullivan L, Vasan RS, Wilson PW, D’Agostino RB (December 2007). “LDL Particle Number and Risk of Future Cardiovascular Disease in the Framingham Offspring Study – Implications for LDL Management”. Journal of Clinical Lipidology. 1 (6): 583–592. doi:10.1016/j.jacl.2007.10.001.
3. Sniderman AD, Lamarche B, Contois JH, de Graaf J (December 2014). “Discordance analysis and the Gordian Knot of LDL and non-HDL cholesterol versus apoB”. Current Opinion in Lipidology. 25 (6): 461–467. doi:10.1097/MOL.0000000000000127.
4. Miller M, Stone NJ, Ballantyne C, Bittner V, Criqui MH, Ginsberg HN, et al. Triglycerides and cardiovascular disease: a scientific statement from the American heart association. Circulation. (2011) 123:2292–333. doi: 10.1161/CIR.0b013e3182160726
5. Chapman MJ, Ginsberg HN, Amarenco P, Andreotti F, Boren J, Catapano AL, et al. Triglyceride-rich lipoproteins and high-density lipoprotein cholesterol in patients at high risk of cardiovascular disease: evidence and guidance for management. Eur Heart J. (2011) 32:1345–61. doi: 10.1016/S1567-5688(11)70033-2
6. Barrows BR, Parks EJ. Contributions of different fatty acid sources to very low-density lipoprotein-triacylglycerol in the fasted and fed states. J Clin Endocrinol Metab. (2006) 91:1446–52. doi: 10.1210/jc.2005-1709
7. Taskinen MR, Adiels M, Westerbacka J, Söderlund S, Kahri J, Lundbom N, et al. Dual metabolic defects are required to produce hypertriglyceridemia in obese subjects. Arterioscler Thromb Vasc Biol. (2011) 31:2144–50. doi: 10.1161/ATVBAHA.111.224808
8. Gaw A, Packard CJ, Lindsay GM, Griffin BA, Caslake MJ, Lorimer AR, et al. Overproduction of small very low density lipoproteins (Sf 20-60) in moderate hypercholesterolemia: relationships between apolipoprotein B kinetics and plasma lipoproteins. J Lipid Res. (1995) 36:158–71.
9. James RW, Martin B, Pometta D, Fruchart JC, Duriez P, Puchois P, et al. Apolipoprotein B metabolismin homozygous familial hypercholesterolemia. J Lipid Res. (1989) 30:159–69.
10. Millar JS, Maugeais C, Ikewaki K, Kolansky DM, Barrett PH, Budreck EC, et al. Complete deficiency of the low-density lipoprotein receptor is associated with increased apolipoprotein B-100 production. Arterioscler Thromb Vasc Biol. (2005) 25:560–5. doi:10.1161/01.ATV.0000155323.18856.a2
11. Björnson E, Packard CJ, Adiels M, Andersson L, Matikainen N, Söderlund S, et al. Apolipoprotein B48 metabolism in chylomicrons and very low-density lipoproteins and its role in triglyceride transport in normo- and hypertriglyceridemic human subjects. J Intern Med. (2019). doi: 10.1111/joim.13017.
12. Taskinen MR, Boren J. New insights into the pathophysiology of dyslipidemia in type 2 diabetes. Atherosclerosis. (2015) 239:483–95. doi: 10.1016/j.atherosclerosis.2015.01.039
13. Malmström R, Packard CJ, Watson TD, Rannikko S, Caslake M, Bedford D, et al. Metabolic basis of hypotriglyceridemic effects of insulin in normal men. Arterioscler Thromb Vasc Biol. (1997) 17:1454–64. doi: 10.1161/01.ATV.17.7.1454