
17th January 2021, Dr Chee L Khoo

The pathogenesis of type 2 diabetes involves insulin resistance, β-cell failure, α-cell over-activity and the loss of the incretins. The brain is responsible for homeostatic control of a broad range of variables upon which our survival depends. Why wouldn’t the brain be involved with glucose control? There is increasing evidence that both the pancreas and the brain are jointly responsible for maintaining glucose within and tight setpoint. It is proposed that impairment of this central control system is fundamental to the pathogenesis of T2D. Let’s review the evidence thus far.
The central (brain) glucose sensor
The brain relies almost exclusively on glucose as a fuel. Its role in ensuring its availability is in keeping with principles of physiology. The French physiologist, Claude Bernard wrote “The stability of the internal environment [milieu intérieur] is the condition for the free and independent life” back in the mid-1980s (1,2). This is the underlying principle of homeostasis. He invoked the role of the brain in glucose homeostasis way back then.
A new proposal
We are going to do things the opposite way. Let’s look at the new proposed model describing the role of the brain in glucose homeostasis. We will then look at what evidence we have for the proposed model (see Figure 1).
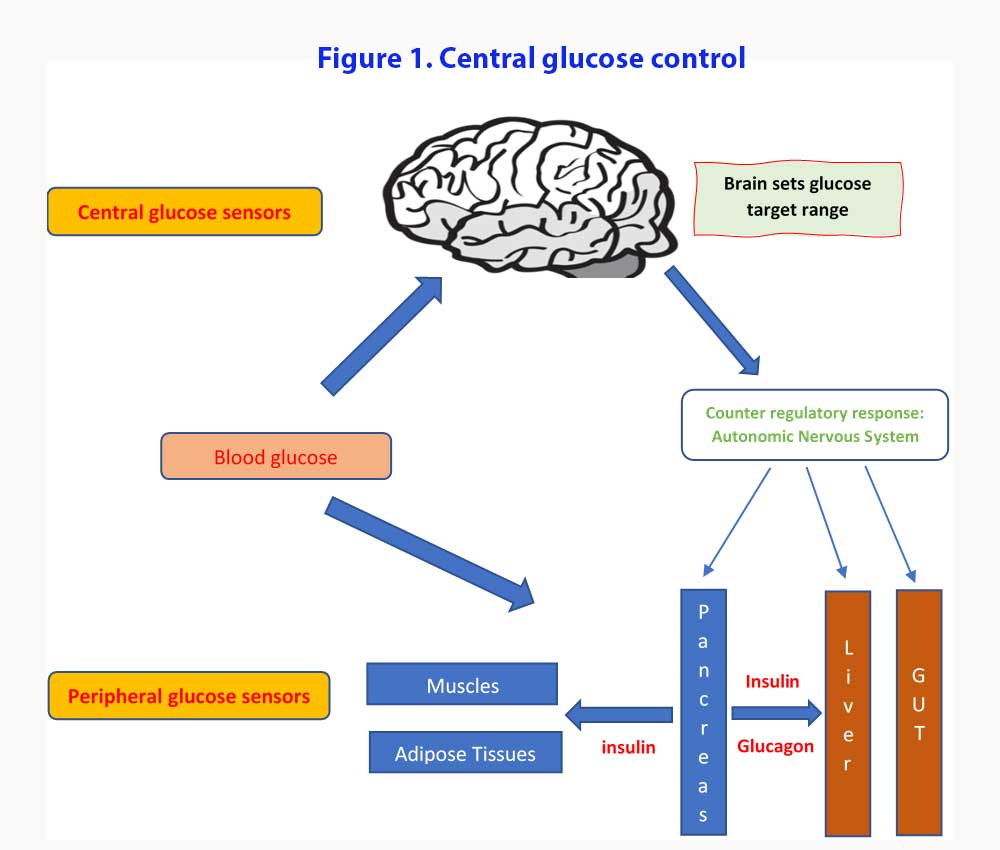
According to this central glucose model, it is the brain that sets up the target glucose set points and work towards maintaining the blood glucose within the range (3). Both peripheral and central glucose-sensing mechanisms monitor the circulating glucose levels and counter regulatory responses (CRR) are enacted when glucose levels stray outside those set targets. Components of the CRR include increased secretion of the hormones glucagon, epinephrine, and glucocorticoids, inhibition of glucose-induced insulin secretion, increased sympathetic nervous system (SNS) outflow to the liver, and increased food intake. Under usual conditions, the pancreatic islets function with a high degree of autonomy with minimal input from the brain while blood glucose levels remain within their defended physiological range [4].
Hypoglycaemia response
Although glucose sensing can occur at peripheral (e.g., neurons innervating the hepatic portal vein) as well as central sites, the brain is the organ responsible both for transducing this information into effective glucose counter regulation and for terminating this response once euglycemia is restored. Of the many brain areas that have been investigated, the hypothalamic ventromedial nucleus (VMN) has emerged as potentially being both necessary and sufficient to elicit this powerful response.
Optogenetics is a biological technique that involves the use of light to control neurons that have been genetically modified to express light-sensitive ion channels. As such, optogenetics uses a combination of techniques from optics and genetics to control the activities of individual neurons in living tissue—even within freely-moving animals. It involves inserting an optic fibre into parts of the brain to activate or deactivate that part of the brain using a light source.
Photo-activation of a specific subset of VMN neurons, VMN steroidogenic factor-1 (VMNSF1) activates the CRR and thereby raises circulating glucose levels (5). Blood glucose levels rise rapidly and markedly, with glycaemia returning to baseline values within 1 h after cessation of photo-stimulation. Glucagon and corticosteroid levels were raised while insulin levels were reduced.
Inhibition of VMNSF1 neurons has no glucose-lowering effect under basal conditions but it severely impairs recovery from hypoglycaemia because it blocks the powerful CRRs that normally restore low blood glucose levels into the normal range. Glucagon and corticosteroid rise were blocked when VMNSF1 “silenced”.
Other researchers were able to trigger glucagon release by inducing neuroglycopaenia with the administration of 2-deoxyglucose locally into the VMN (6). Conversely, the glucagon response to systemic hypoglycaemia is blocked by administration of glucose into the VMN and in rats with bilateral VMH lesions.
Thus, the VMNSF1 neurons are involved in CRRs via a combination of increased CRR hormone secretion and inhibition of glucose-induced insulin secretion.
CNS-Islet relationship
While we think of insulin secretion being a function of peripheral glucose sensors triggering pancreatic beta cells to release insulin, there is ample evidence demonstrating conclusively that release of
insulin (and glucagon) is influenced by neural input under physiological conditions (7, 8). VMNSF1 project to the islet via multi-synaptic relays involving both limbs of the autonomic nervous system (i.e. the sympathetic (SNS) and parasympathetic systems (PNS)). The activity of SNS or PNS fibres influences secretion of both insulin and glucagon in ways that can potently impact blood glucose levels.
Post prandial burst of glucose leads to an increase of PNS outflow to the pancreas leading to increase in insulin secretion. Neural control of islet function also plays a physiological role in cephalic-phase insulin release (insulin secretion in response to feeding cues but before nutrient absorption or increase in blood glucose) (9). This cephalic phase is mediated by vagal cholinergic signals (10).
On the other hand, hypoglycaemia increases SNS outflow to the islet which stimulates glucagon secretion and inhibits insulin secretion. Sympathetic fibres supplying the liver are also activated as part of the CRR and, together with increased plasma adrenaline (arising from activation of the adrenal medulla) and glucagon, these responses drive increased hepatic glucose production in an effort to restore normo-glycaemia (11). Suppression of insulin secretion in this setting involves activation of β-adrenergic receptors on beta cells resulting from either increased SNS outflow directly to the islet or increased circulating adrenaline levels (or both) This glucagon response is impaired if islets are denervated (e.g. islet/pancreas transplantation or diabetic neuropathy) (12).
Another illustration of the role of the brain in glucose control involved intracerebroventricular (i.c.v.) administration of the leptin to rats or mice with uncontrolled, insulin-deficient diabetes (13). The i.c.v. administration of leptin normalised both the excessive hepatic glucose production and the reduced tissue glucose utilisation characteristic of uncontrolled diabetes, despite persistent, severe insulin deficiency. The dose needed to achieve this effect was low enough to have no measurable effect when given systemically.
So, what happens in T2D?
In T2D, the glucose set point is set higher and brain is geared to defend the perceived glucose deficiency by invoking CRRs to defend that higher glucose level. In a recent study by Flak and colleagues (14), silencing a subset of glutamatergic CRR neurons in the VMN of otherwise normal mice caused a ~25% reduction in blood glucose levels. In diabetic mice, inactivation of the VMNSF1 neurons significantly reduce hyperglycaemia.
Now, there is no doubt that failure in insulin secretion due to beta cell dysfunction or death is fundamental to the pathogenesis of T2D. However, beta cell dysfunction predates the development of hyperglycaemia. This is one of those chicken and egg situation. Is beta cell dysfunction the primary, initial defect or is the dysfunction the result of an interaction between genetic susceptibility and metabolic consequences of the disease (e.g. ‘glucotoxicity’ and ‘lipotoxicity’)?
Perhaps, in genetically susceptible individuals, any pathological process that raises the defended level of blood glucose (including defective brain sensing) triggers the vicious cycle of escalating beta cell dysfunction and metabolic impairment characteristic of type 2 diabetes. A brain mechanism that drives hyperglycaemia in part by inhibiting insulin secretion when VMNSF1 neurons are activated may be what set this pathological cascade in motion.
Full understanding of the brain’s role in both glucose homeostasis and type 2 diabetes pathogenesis remains in its infancy. Further understanding of this brain glucose control can inform type 2 diabetes treatment strategies in the future which can restore the defended level of blood glucose to normal even in patients with limited islet cell reserve.
References:
- https://en.wikipedia.org/wiki/Claude_Bernard. Accessed 16th January 2021
- Bernard C (1849) Chiens rendus diabetiques. C R Soc Bio 1:60
- Alonge KM, D’Alessio DA, Schwartz MW. Brain control of blood glucose levels: implications for the pathogenesis of type 2 diabetes. Diabetologia. 2021 Jan;64(1):5-14. doi: 10.1007/s00125-020-05293-3. Epub 2020 Oct 12. Erratum in: Diabetologia. 2020 Nov 11;: PMID: 33043401; PMCID: PMC7718404.
- Schwartz MW, Seeley RJ, Tschöp MH et al (2013) Cooperation between brain and islet in glucose homeostasis and diabetes. Nature 503(7474):59–66. https://doi.org/10.1038/nature12709
- Meek TH, Nelson JT, Matsen ME, Dorfman MD, Guyenet SJ, Damian V, Allison MB, Scarlett JM, Nguyen HT, Thaler JP, Olson DP, Myers MG Jr, Schwartz MW, Morton GJ. Functional identification of a neurocircuit regulating blood glucose. Proc Natl Acad Sci U S A. 2016 Apr 5;113(14):E2073-82. doi: 10.1073/pnas.1521160113. Epub 2016 Mar 21. PMID: 27001850; PMCID: PMC4833243.
- Asplin CM, Raghu PK, Koerker DJ, Palmer JP (1985) Glucose counterregulation during recovery from neuroglucopenia: which mechanism is important? Metabolism 34(1):15–18. https://doi.org/10.1016/0026-0495(85)90053-8
- Woods SC, Porte D Jr (1974) Neural control of the endocrine pancreas. Physiol Rev 54(3):596–619. https://doi.org/10.1152/physrev.1974.54.3.596
- OsundijiMA, EvansML (2013) Brain control of insulin and glucagon secretion. Endocrinol Metab Clin N Am 42(1):1–14. https://doi.org/10.1016/j.ecl.2012.11.006
- Teff KL (2011) How neural mediation of anticipatory and compensatory insulin release helps us tolerate food. Physiol Behav 103(1):44–50. https://doi.org/10.1016/j.physbeh.2011.01.012
- Thorens B (2014) Neural regulation of pancreatic islet cell mass and function. Diabetes Obes Metab 16(Suppl 1):87–95. https://doi.org/10.1111/dom.12346
- Perseghin G, Regalia E, Battezzati A et al (1997) Regulation of glucose homeostasis in humans with denervated livers. J Clin Invest 100(4):931–941. https://doi.org/10.1172/jci119609
- Taborsky GJ Jr, Mundinger TO (2012) Minireview: the role of the autonomic nervous system in mediating the glucagon response to hypoglycemia. Endocrinology 153(3):1055–1062. https://doi.org/10.1210/en.2011-2040
- German JP, Thaler JP, Wisse BE et al (2011) Leptin activates a novel CNS mechanism for insulin-independent normalization of severe diabetic hyperglycemia. Endocrinology 152(2):394–404. https://doi.org/10.1210/en.2010-0890
- Flak JN, Goforth PB, Dell Orco J et al (2020) Ventromedial hypothalamic nucleus neuronal subset regulates blood glucose independently of insulin. J Clin Invest 130(6):2943–2952. https://doi.org/10.1172/jci134135